Scanning for Recent Human Evolution
![]() Survival of the fittest is a concept that is well known to most of us. Heaven knows, many of us strive to remain fit enough to try and extend our life expectancy and survive. But in its original context this relates to natural selection and evolution. A lot of the time natural selection is a conservative force (“negative selection“), trying to keep an organism close to a previously achieved state of perfection. However, occasionally circumstances can change, and selection can then favour genetic changes that will fit an organism better for the new regime. This is what we call “positive selection“, and unlike its rather boring conservative cousin, it can be very interesting indeed.
Survival of the fittest is a concept that is well known to most of us. Heaven knows, many of us strive to remain fit enough to try and extend our life expectancy and survive. But in its original context this relates to natural selection and evolution. A lot of the time natural selection is a conservative force (“negative selection“), trying to keep an organism close to a previously achieved state of perfection. However, occasionally circumstances can change, and selection can then favour genetic changes that will fit an organism better for the new regime. This is what we call “positive selection“, and unlike its rather boring conservative cousin, it can be very interesting indeed.
Continuing the unveiling of our top ten papers selected from the papers published in PLOS Biology over the last decade, the latest choice morsel comes from 2006. The authors developed a new statistical method to search for features that flag those regions of the human genome that may have aided our own adaptation to changing fortunes. They applied this method to the then recently available single nucleotide polymorphism (SNP) data from the International HapMap Project – a catalogue of subtle (but potentially important) genetic variations in different human populations.
![By TimVickers at en.wikipedia [Public domain], from Wikimedia Commons The source of this image is the frontispiece of Huxley's 1863 book Man's Place in Nature.](https://biologue.plos.org/wp-content/uploads/sites/7/2020/05/Huxley_-_Mans_Place_in_Nature-2.png)
The source of this image is the frontispiece of Huxley’s 1863 book Man’s Place in Nature.
Senior author of the study, Jonathan Pritchard, commented that “this was an important paper for my lab. It really represented the start of our transition into thinking about genome-wide variation data.” He notes also that “[this project] brought me back to thinking about how positive selection shapes the genome… the first area that excited me when I got into biology.” The study was groundbreaking. It presented both the authors’ new statistical method to scan the genome, and many intriguing insights about what had been (and is still being) selected for in humans.
I fully intended to describe the paper to you in some detail, but luckily for me long-time Editorial Board member, and the academic editor who advised us through the review process back then, Laurence Hurst, remembers it well:
“I recall that this paper came at an interesting time. Plenty of folks were attempting between-species trawls for domains under selection (using Ka/Ks etc) but this was one of the first to consider genome-wide trawls for selection based on SNP/haplotype data and so capture much more recent selection events – potentially events that reflect different selection pressures in different populations.”
He goes on to note that the author came up with “a simple but elegant solution” and that “all methods of this variety also have problems with demography (which can give false signals), but the paper makes a good crack at doing simulations to try and weed out these issues.”
“I liked this study,” continues Hurst, “not least because it is novel, clever, rigorous and careful, but it is interesting to see what selection is doing. I find it intriguing, for example, that skin pigment genes appear to be under selection in Europeans. But this isn’t just academic interest: as the authors note, alleles under recent selection are often associated with complex phenotypes of medical relevance. Indeed they identify alleles associated with alcohol susceptibility and salt-sensitive hypertension.”
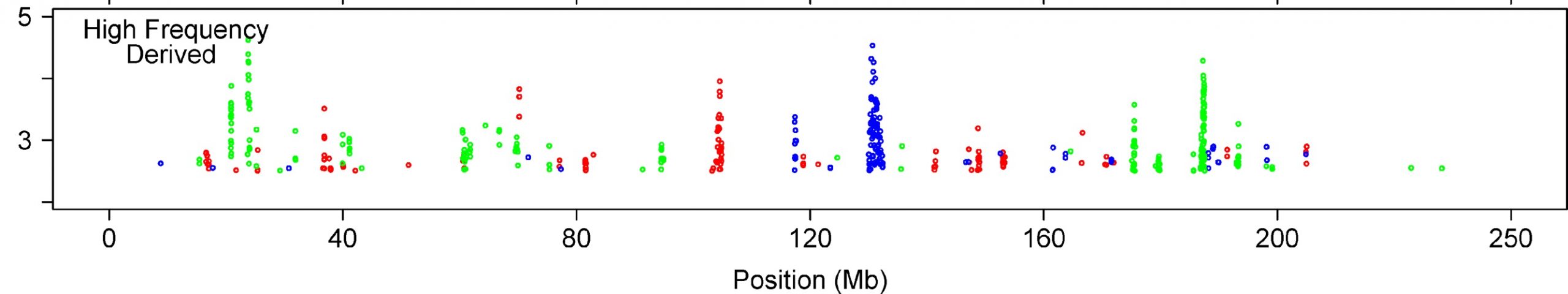
In short, the authors compared SNP data from three populations – Yorubans from sub-Saharan Africa, a combination of Japanese and Chinese individuals from Asia, and a cohort from Europe. They specifically looked across the genome for long blocks of co-inherited DNA (haplotypes) that indicate that a particular SNP (or a nearby feature in the genome) confers some positive advantage on the individual that carries it, and that it is increasing in prevalence in that population. Their new method was a breath of fresh air, and has been widely adopted since then, and they found lots of intriguing biologically and medically relevant results. What’s not to like?
PLOS Biology editorial board member Chris Tyler-Smith noted that this is “A classic paper in my own field, introducing a method that has now become standard”. One of the two co-first authors, Ben Voight, was a graduate student when undertaking this research and is now an Assistant Professor at U. Penn. Voight notes that this paper probably “was a non-trivial factor in getting me where I am today”. In recalling his work on this study, he remembers that given the competitive nature of the field at that time and as a newcomer they were very careful in “crafting the description of the framework, model, and inferences possible. This took a great deal of effort, but in some way was only really possible by a publication framework where we weren’t unnecessarily limited in making that description”.
And it seems that this area is rife for future additional discovery as Pritchard muses “More recently my lab [and others] has argued that adaptation by polygenic selection is likely much more important than standard sweep models… in the next few years it will be possible to make a lot of progress on untangling these issues, helped … by the much richer sequence data that are available today”. Voight told me that “it is clear that this work opened the door to a number of new questions which follow that, even today, have not been flushed out completely: the times these sweeps occurred in human history, the targets, mechanisms, the phenotypes subject to fitness consequences, and the relationship to complex traits and diseases.” And so as our resources and tools improve and advance, our ability to decipher and decode backwards progresses in tandem, leading us to better understand how we came to be who, what, and where we are today. The fascinating research will continue…
If you want to read more about the original research article, we published both a synopsis and an editorial in the same issue of the journal way back when.
![]() See the Tenth Anniversary PLOS Biology Collection or read the Biologue blog posts highlighting the rest of our selected articles.
See the Tenth Anniversary PLOS Biology Collection or read the Biologue blog posts highlighting the rest of our selected articles.

Voight BF, Kudaravalli S, Wen X, & Pritchard JK (2006). A map of recent positive selection in the human genome. PLoS biology, 4 (3) PMID: 16494531